中国经济网北京12月14日讯昨日,君实生物(688180.SH)发布公告称,特瑞普利单抗联合化疗一线治疗非小细胞肺癌的III期临床研究达到主要研究终点。 公告显示,近日,君实生物产品特瑞普利单抗(商品名:拓益?,产品代号:JS001)联合化疗一线治疗晚期非小细胞肺癌的随机、双盲、多中心的III期临床研究(以下简称“CHOICE-01研究”,Clinicaltrial.gov登记号:NCT03856411)在期中分析中,由独立数据监察委员会(IDMC)判定达到了预设的主要研究终点。公司将于近期向国家药品监督管理局(以下简称“国家药监局”)递交新适应症上市申请。 关于药品的基本情况,特瑞普利单抗是中国首个批准上市的以PD-1为靶点的国产单抗药物。特瑞普利单抗自2016年初开始临床研发,至今已在中、美等多国开展了覆盖超过15个瘤种的30多项临床研究。2018年12月17日,特瑞普利单抗获得国家药监局有条件批准上市,用于既往接受全身系统治疗失败的不可切除或转移性黑色素瘤的治疗。特瑞普利单抗用于三线治疗复发╱转移性鼻咽癌,以及特瑞普利单抗用于二线治疗转移性尿路上皮癌的新适应症上市申请分别于2020年4月和2020年5月获得国家药监局受理。2020年7月,上述两项新适应症上市申请已于被国家药监局纳入优先审评程序。2020年9月,特瑞普利单抗用于治疗复发╱转移性鼻咽癌获得美国食品药品监督管理局突破性疗法认定。 关于CHOICE-01研究,CHOICE-01研究是一项随机、双盲、多中心的III期临床研究,旨在评估特瑞普利单抗联合标准一线化疗对比安慰剂联合化疗,在未经治疗的晚期鳞状和非鳞状非小细胞肺癌患者中一线治疗的有效性与安全性。该研究由中国医学科学院肿瘤医院王洁教授担任主要研究者。 CHOICE-01研究是国内首个将抗PD-1单抗联合化疗作为一线治疗,在晚期鳞状和非鳞状非小细胞肺癌两种组织学亚型患者中开展的III期临床研究。CHOICE-01研究实际入组465例非小细胞肺癌患者,其中鳞状非小细胞肺癌患者220例,非鳞状非小细胞肺癌患者245例。该研究的主要研究终点为无进展生存期(PFS),次要研究终点为总生存期(OS)、客观缓解率(ORR)、缓解持续时间(DOR)、疾病控制率(DCR)以及安全性等。 君实生物表示,根据CHOICE-01研究期中分析结果,独立数据监察委员会(IDMC)判定该研究主要研究终点无进展生存期达到方案预设的优效界值,特瑞普利单抗安全性数据与已知风险相符,未发现新的安全性信号。根据我国药品注册相关的法律法规要求,药物需完成临床研究并经国家药监局审评、审批通过后方可生产上市。 君实生物在公告中强调,由于医药产品具有高科技、高风险、高附加值的特点,药品的前期研发以及产品从研制、临床试验报批到投产的周期长、环节多,容易受到一些不确定性因素的影响。敬请广大投资者谨慎决策,注意防范投资风险。 今日,君实生物股价上涨,截至收盘报67.01元,涨幅2.76%,成交额9142.85万元,换手率1.95%。值得注意的是,君实生物此前已连续5个交易日股价下跌,12月7日收盘跌0.53%,12月8日收盘跌1.55%,12月9日收盘跌2.69%,12月10月收盘跌0.65%,12月11日收盘跌3.61%。
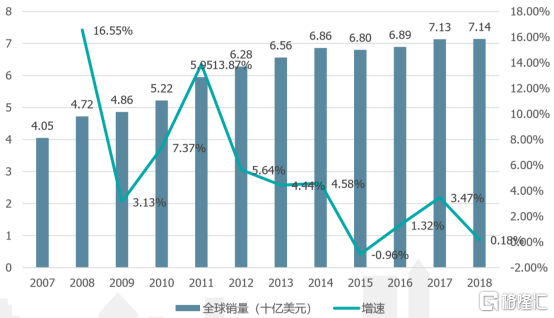
近日,复宏汉霖-B(2696.HK)旗下的曲妥珠单抗汉曲优®(HLX02,欧盟商品名:Zercepac®)正式获得国家药品监督管理局(NMPA)批准上市,成为首个中欧两地获批上市的国产单抗生物类似药。汉曲优®的获批实现了国产曲妥珠零的突破,打破曲妥珠单抗自2002年国内上市以来长期只有单一品种在市销售的局面,有望改变国内抗HER2治疗格局。同时,作为首个登陆欧洲市场的“中国籍”单抗生物类似药,汉曲优®更开辟了中国医药企业参与单抗生物类似药“世界杯”比赛的先河。 国产替代与国际竞争并重 在中国临床肿瘤学会发布的《乳腺癌诊疗指南》中,曲妥珠单抗联合化疗被推荐为HER2阳性乳腺癌术前治疗方案(I级推荐)、HER2阳性早期乳腺癌术后辅助靶向治疗(I级推荐)以及 HER2 阳性晚期(复发转移)乳腺癌的抗 HER2 一线和二线治疗方案(I级推荐)。曲妥珠单抗在乳腺癌全程治疗中起到了奠基石的作用。然而长期以来在中国上市的曲妥珠单抗仅有国外药企罗氏的赫赛汀®,自该药2002年上市以来已独家占据市场长达 18 年。 在这样的局面下,进口药物费用昂贵,年度治疗费用高达10万,给患者家庭带来沉重的用药负担;随着该药被纳入医保目录,又一度出现医保报销范围内药物供应紧张,部分地区无法及时调配药物的局面。医生、患者与市场都期待出现新的破局者,能够提供价格可及的优质药物,稳定持续满足日渐增多的乳腺癌患者的用药需求。 根据官方发布的临床试验(1期与国际多中心3期)结果,复宏汉霖自主开发与生产的汉曲优®(HLX02)与原研曲妥珠在药代动力学特征、安全性、耐受性和免疫原性上皆相似,两者无临床意义上的差异。此次汉曲优®也获批了赫赛汀®在中国已获准的全部适应症,使得中国HER2阳性乳腺癌和胃癌患者终于增加了新的曲妥珠单抗的用药选择。 不仅于此,2020年4月,HLX02曲妥珠单抗原液(DS)和制剂(DP)线顺利通过欧盟GMP现场核查,正式获得了两项欧盟GMP证书。欧盟GMP认证是国际公认的最权威和严谨的认证之一,欧盟GMP认证被也视为药品登陆国际市场的“通行证”。一个多月后,欧洲药品管理局EMA发布积极审评意见,表示“HLX02与参照药赫赛汀(曲妥珠单抗)高度相似,研究数据支持HLX02在质量、安全性与疗效等方面与赫赛汀均无显著差异”,建议批准HLX02的上市销售许可申请。2020年7月27日,欧盟委员会同意HLX02(曲妥珠单抗,欧盟商品名:Zercepac®)在欧盟上市,适应症范围包括HER2阳性早期乳腺、HER2阳性转移性乳腺癌以及HER2阳性转移性胃癌,即赫赛汀®已获准的全部适应症。汉曲优®由此成为首个登陆欧洲市场的“中国籍”单抗生物类似药,开辟了中国医药企业参与单抗生物类似药“世界杯”比赛的先河。 需求与供应,现实与未来 曲妥珠单抗(赫赛汀®)于1998年在美国上市,是HER2阳性乳腺癌治疗的“金标准”药物。经过了20年发展,曲妥珠单抗2018年全球销售额仍超过70亿美元,排药品世界销售额第七位。而乳腺癌在2018年发病率位居全球恶性肿瘤第二位,同时位居女性恶性肿瘤第一位,在癌症患者基数持续上涨的背景下,曲妥珠单抗的需求持续强劲,广大患者群体对高品质生物类似药需求旺盛。 图表 1:赫赛汀全球历年销售额 资料来源:Bloomberg,兴业证券经济与金融研究院,整理 自2002年上市以来,曲妥珠单抗在中国的销售额稳步上升,销售额上升的主要限制在于能够负担药物开支的患者群体有限。自2017年曲妥珠单抗加入我国医保后药品使用总量急剧上升,但需求的急剧释放也导致了药品供应不足的等问题。 图表 2:曲妥珠单抗国内销售额 资料来源:兴业证券经济与金融研究院,整理 综上所述,曲妥珠单抗目前在国内处于需求旺盛,但供应不稳定的局面。该局面下复宏汉霖开发的国产生物类似药凭借与原研一致的疗效和国际质量品质,将有充足的市场空间——汉曲优®有望利用本土生产的优势保持充足供货,填补国内用药需求,同时走出国门,参与分配国内外市场份额,代表中国本土生物药打世界杯。 据悉,复宏汉霖已为产品的全球化产能需求作好了充分准备。公司已将现有11000平方米的徐汇生物药生产基地的商业化产能扩至20,000L,并同步启动松江基地(一)与松江基地(二)的建设。松江基地(一)预计产能达24,000L,现已完成临床样品的试生产,松江基地(二)总占地面积达到约200亩,一期智能化工厂也正在建设中。 市场关注与赞同 复宏汉霖是中国抗体药龙头企业之一,在单抗生物药的研发和国际化方面进度领先,综合竞争实力强劲。基于以上客观条件,多家外部券商给予复宏汉霖积极的评级。招银国际维持“买入”评级,目标价57.21港元,花旗首次给与“买入”评级,目标价58港元,光大证券首次给与“买入”评级,目标价64港元。随着外部逐渐发掘企业投资价值,市场关注度也在不断上升,复宏汉霖(2696.HK)自6月12日开始成交量逐步放大,6月16日单日涨幅超过14%,短期盘整之后7月10日开启新的一轮放量上涨,四个交易日实现25%的涨幅。放量资金的进入也是市场对企业潜在盈利预期的认可,关注度上升,市值上涨,产品投产后转化为实际业绩支撑市值,促使业绩与估值成为一个正向反馈的循环。 小结 随着本次HLX02汉曲优®的在国内及欧盟地区双双获批,复宏汉霖实现了国产曲妥珠单抗零的突破,并使得“中国籍”单抗生物类似药首次登陆欧洲市场。该产品优异的质量和严格的开发、生产过程使得其与原研药在质量、有效性与安全性上达到了高度相似,将为复宏汉霖打开国内外这一生物医药主流市场的大门。凭借全球对曲妥珠单抗的旺盛需求,复宏汉霖立足国内、布局全球,有望充分发挥本土生产优势占据市场,并凭借稳定的生产工艺赢得国际认可打开国际销售之路。在这商业化现实条件充分,企业国内外盈利前景明朗的市场背景下,复宏汉霖有望走好国内国产替代、国外优势竞争的“内外”兼修之路。
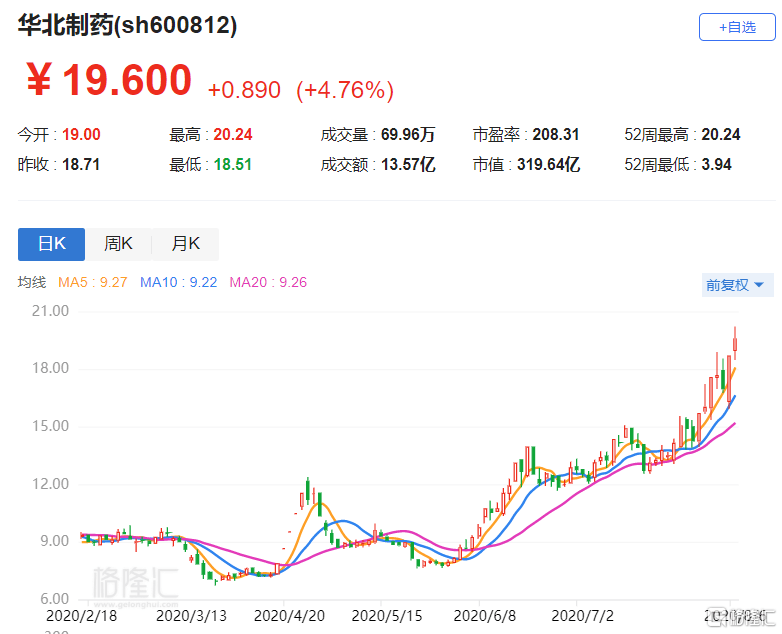
8月6日丨华北制药(600812.SH)涨4.76%,报19.6元,股价再创上市以来新高,总市值319.6亿元。 华北制药昨晚公告,公司子公司新药公司的重组人源抗狂犬病毒单抗注射液8月5日被国家药品审评中心纳入拟优先审评品种公示名单,公示期7日。重组人源抗狂犬病毒单抗注射液(rhRIG)是新药公司自主创新项目,被列为国家“重大新药创制”科技重大专项品种。目前,国内尚无重组抗狂犬病毒单抗药物上市销售。
8月5日晚间,华北制药发布公告称,公司子公司华北制药集团新药研究开发有限责任公司(以下简称“新药公司”)的重组人源抗狂犬病毒单抗注射液2020年8月5日被国家药品监督管理局药品审评中心(以下简称“国家药品审评中心”)纳入拟优先审评品种公示名单,公示期7日。 公告显示,拟优先审评理由:经审核,本申请符合新《药品注册管理办法》规定的优先审评审批范围,同意按优先审评审批程序第六十八条:(一)临床急需的短缺药品、防治重大传染病和罕见病等疾病的创新药和改良型新药纳入优先审评程序。 重组人源抗狂犬病毒单抗注射液(rhRIG)是新药公司自主创新项目,被列为国家“重大新药创制”科技重大专项品种。其作用机制及适应症为将本品与人用狂犬病疫苗联用,用以补充人用狂犬病疫苗主动免疫过程中的抗体空白,可直接中和体内狂犬病毒,起到被动免疫作用,用于被狂犬或其它狂犬病毒易感动物咬伤、抓伤患者的被动免疫。 2020年4月7日,该项目取得III期临床试验报告,试验结论为:重组人源抗狂犬病毒单抗注射液(rhRIG)联合人用狂犬病疫苗对III级疑似狂犬病毒暴露人群的暴露后预防达到主要疗效和次要疗效终点,且安全性良好,达到方案设定目标,试验药物重组人源抗狂犬病毒单抗注射液(rhRIG)是安全、有效的。 2020年6月26日,国家药品审评中心受理了重组人源抗狂犬病毒单抗注射液药品注册申请。 公告显示,目前,国内尚无重组抗狂犬病毒单抗药物上市销售。国内可用于狂犬病毒暴露后预防的被动免疫制剂为抗狂犬病血清和狂犬病人免疫球蛋白。 通过国家药品监督管理局网站数据查询结果显示,截至目前国内有17家企业具有狂犬病人免疫球蛋白批准文号,未批准进口产品销售。 根据Insight数据库统计的中检院及七家药品检验所公开的生物制品批签发数据,2019年批签发狂犬病人免疫球蛋白1216.6万瓶(折合200IU)。根据药智网药品中标信息查询,狂犬病人免疫球蛋白2019年各地中标价为143.3元-218元(2ml/200IU)不等。 截至目前,该项目累计研发投入金额为1.64亿元。





 登录
登录



 工商服务
工商服务 危机公关
危机公关 金融律师
金融律师 app开发
app开发 财税服务
财税服务 金融牌照
金融牌照 网站建设
网站建设 知识产权
知识产权 企业征信
企业征信 审计评估
审计评估